
Coupling of reactive and non-reactive potentials for the study of cracking by molecular simulation
Research team: L. Brochard and F. Legoll (Navier), E. Cances, T. Lelièvre and G. Stoltz (CERMICS)
December 2013 – November 2014
The one-year postdoc of Ignacio Tejada was dedicated to the development of an original methodology to improve the efficiency of molecular simulations involving reactive potentials. Reactive potentials are a specific category of inter-atomic potentials used in classical molecular simulations (molecular dynamics, Monte Carlo) that have the ability to adapt to the atomic environment. Therefore these potentials are suited for the study of phenomena involving rearrangements at the atomic scale, for instance failure of materials. However, those potentials have a computational cost one to two orders of magnitude higher than conventional non-reactive potentials. Improving performance is much needed for practical applications, especially in the field of failure mechanics for which typical molecular simulations involves about 104 to 106 atoms and can last weeks on supercomputers. One way of improving the performance, which was the starting point in this project, is to limit the use of the reactive potential to small regions of the studied system where the reactive ability is required. Indeed, very often the reactive ability is useful only in some small regions of a molecular simulation, e.g., crack tips in simulation of fracture, solid surface for simulation of surface catalysis etc. Accordingly, the methodology we developed in this project aimed at substituting the reactive potential by a non-reactive counterpart wherever possible. Development of the methodology involved: 1- the approach to construct the non-reactive counterpart, 2- the approach to couple the reactive potential and its non-reactive counterpart in a seamless manner, 3- the construction of criterion allowing for on the fly substitution in a simulation. Molecular simulations of graphene failure with the Reactive Bond Order (REBO) were considered as an example of application of the proposed methodology. For this particular example, the improvement in computational cost was about a factor of 5. The computational cost of REBO being one of the smallest among reactive potentials, the potential improvement for other reactive potential could be even higher. An illustration of the methodology is provided in the figures below.
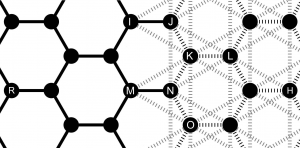

When the reactivity of the potential is unnecessary, the reactive potential is replaced by a non-reactive counterpart made of a set of harmonic springs and angles between first- and second-nearest neighbors. The non-reactive approximation reproduces exactly the Hessian of the reactive potential, which makes it suitable for substitution at small strains and temperatures. A bond-by-bond substitution is used (one reactive bond replaced by a set of springs and angles) which is particularly convenient for on-the-fly adaptive coupling. Finally, special care is needed at the interfaces between the reactive and non-reactive potentials to ensure a seamless coupling.
A numerical code was developed to implement the methodology in the case of 2D-graphene systems modeled with REBO. This code was used to study the failure behavior of graphene-like system with flaws (cracks, holes) in order to investigate of the long-standing issue of failure initiation in a flawed material: for graphene sheets much larger than 3.5 nm, one can distinguish between energy-driven failure (fracture failure) and stress-driven failure (yield failure). Simulations have been performed on flawed graphene sheets of 10 to 20 nm to investigate the detailed transition from fracture to yield failure, which strongly benefited from the methodology established in the postdoc project. The results obtained were particularly interesting since it appeared that only one of the many initiation theories (Leguillon’s criterion) could capture the failure behavior predicted for these systems. This finding raises interesting questions about the physics of failure initiation and is the focus of another Labex project (PhD of Sabri Souguir).
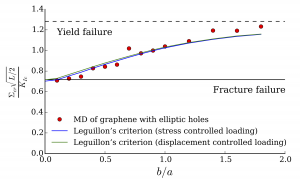
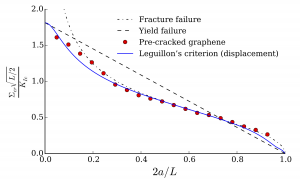
Publications in international journals
[1] Tejada, I. G., Brochard, L., Stoltz, G., Legoll, F., Lelièvre, T. and Cancès, E. (2015), Combining a reactive potential with a harmonic approximation for molecular dynamics simulation of failure: construction of a reduced potential, Journal of Physics: Conference Series, 574, 012041. https://hal.archives-ouvertes.fr/hal-01095102
[2] Tejada, I. G., Brochard, L., Lelièvre, T., Stoltz, G., Legoll, F. and Cancès, E. (2016), Coupling a reactive potential with a harmonic approximation for atomistic simulations of material failure, Computer Methods in Applied Mechanics and Engineering, 305, 422–440. https://hal.archives-ouvertes.fr/hal-01196505
[3] Brochard, L., Tejada, I. G. and Sab, K. (2016), From yield to fracture, failure initiation captured by molecular simulation, Journal of the Mechanics and Physics of Solids, 95, 632–646. https://hal.archives-ouvertes.fr/hal-01686201
Ignacio Tejada is currently assistant professor, Universidad Politécnica de Madrid, Departamento de Ingeniería y Morfología del Terreno