
Using DFT calculations with van der Waals correction, we have systematically investigated the structure and the stability of carbon clusters, single- and bi-layer organic molecules on the Cu(111) surface. In the first part, the interaction between two types of stable carbon clusters (C21 and C24) and the Cu(111) surface in different adsorption sites and rotation angles have been studied using VASP[1] and PBE[2]. These clusters adopt a domelike shape after adsorption on Cu substrate due to the exclusive bonding of the peripheral carbon atoms to the Cu substrate which confirms that a cluster edge tends to bond chemically to the Cu surface. It is found that the binding energy of C21@Cu(111) is at least twice greater than the binding energy of C24@Cu(111). For all the studied adsorption conformations, the PBE binding energies are slightly larger than those obtained with vdW functionals which confirms that vdW interactions play a minor role in these systems.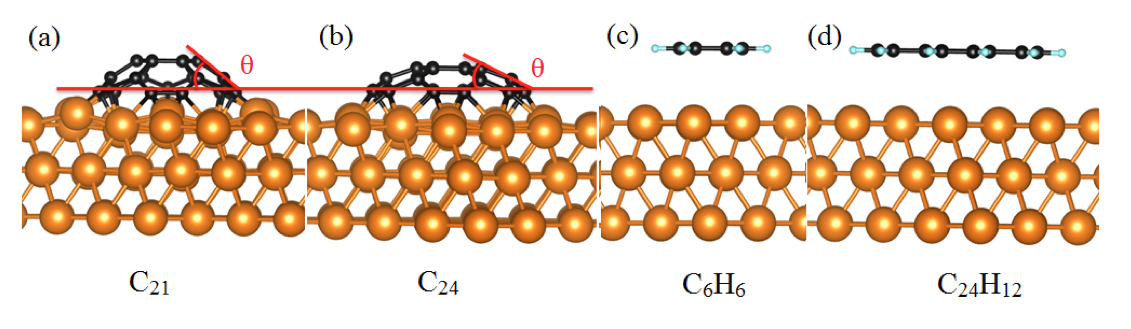
Fig 1. The side view of relaxed geometries for (a) C21, (b) C24, (C) benzene (C6H6) and (d) coronene (C24H12) absorbed on Cu (111) surface.
Then, the adsorption of the single- and bi-layer benzene and coronene molecules on Cu(111) have been examined. In these cases, the long-range vdW forces are important for understanding the interaction between organic molecules and Cu surface. The presence of the edge H atoms maskes the chemical reactivity of the edge C atoms. The binding energies for the organic molecules on Cu(111) systems are significantly smaller than those for C21 and C24 clusters.
The present vdW-DF [3] and vdW-DF2 [4] results demonstrate that the FCC0° site is a more stable adsorption site for benzene/Cu(111), while the HCP30° site is more stable than any other structures for coronene/Cu(111). The most stable conformation for bi-layer benzene and coronene molecules is the AB0° stacking which has the shortest interlayer distance. Charge density difference and binding energy analyses suggest that the adsorption strength would decreases with the increase of PAH sizes on Cu(111). In addition, we have shown that there is a positive linear dependence between the binding energy per carbon atom and the ratio NH/NC for adsorbed single- and bi-layer PAH molecules. We have also found a positive linear dependence between the amount of electronic charge transferred from the PAH to the Cu surface which is consistent with the behavior displayed by the binding energies.
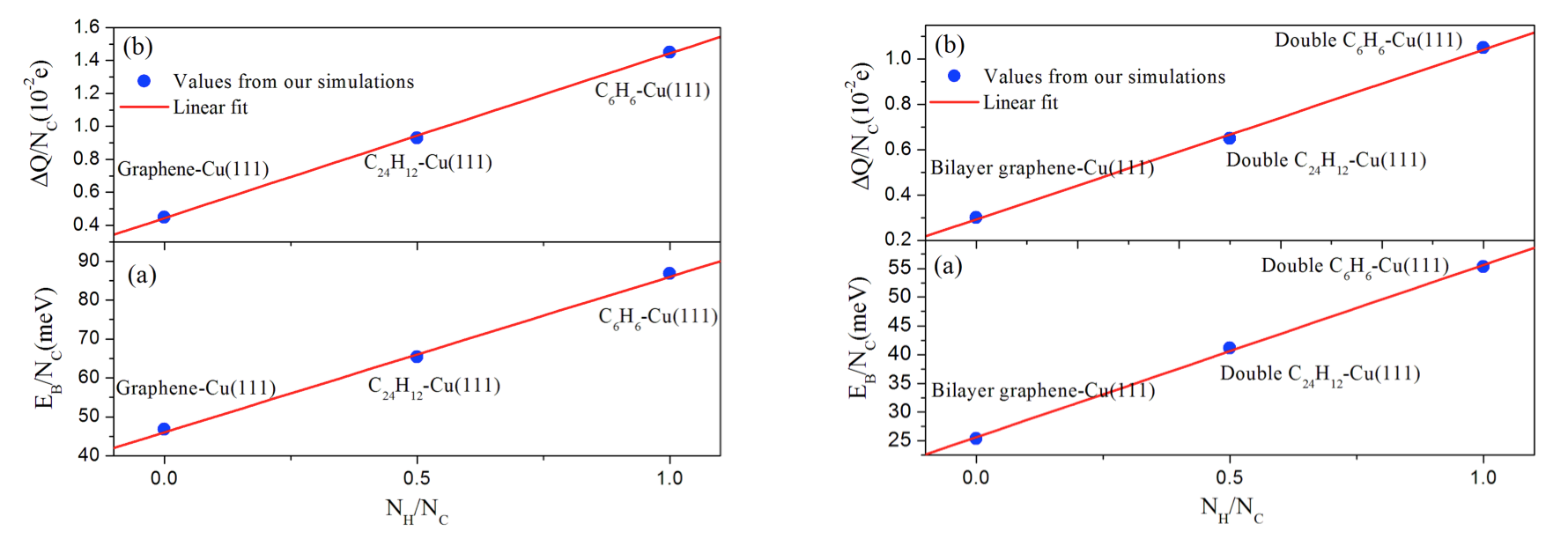
Fig. 2. Calculated and fitted values for the (a) binding energies and (b) charge transfers (per C atom) for the single benzene, single coronene and monolayer graphene (left hand side) and for the bi-layer benzene, coronene and graphene wall (right hand side) on Cu(111) as a function of the NH /NC ratio. NH and NC correspond to the number of hydrogen and carbon atoms, respectively.
During the present study, several controversies appeared concerning the use of vdW-DF2 despite previous successful results on Ar@Au(111) [5]. Some alternatives, such as the approach developed by Tkatchenko and collaborators [6] or PBE-dDsC [7], are still investigated.
In the point of view the passivation, the main conclusions that already can be derived are the followings:
- The vdW-DF functional should be used to study edges with H atoms and PBE for edges with only C atoms.
- The H atom edges lead to physisorption effects such as small oxydative molecules could insert between the graphene and the Cu material.
- For carbon edges, the alternation of pentagons and hexagons leads to stronger binding to the Cu surface. Thus, the defects of this structure would be expected to be more stable. Oxydative molecule are probably stronger blocked by this interaction, in such way that their insertion between the graphene and Cu should be impossible.
- The orientation effects play only a minor role.
- The addition of a second layer decreases the interaction of the carbon system with the Cu slab such that more oxydative molecules could pass inside the interface. The use of monolayer graphene is then more appropriate to avoid these drawbacks.
Additional first-principle investigations should be performed on graphene with defects in presence of oxydative molecules such that O2 in order to examine these points. Then, a homogenization approach, with a description of the defects at a large scale, must follow to study the mechanical properties of the whole material, exposed or not to oxidation in a future work.
In parallel to this main study, some interesting electronic and optical properties of 2D-slab, such as BeO or graphene doped by Si atoms, have been investigated in the continuity of the previous works of Dr Shahrokhi. Two publications have been published in Journal of Alloys and Compounds.
References:
[1] G. Kresse, J. Furthmuller, Phys. Rev. B 54 (1996) 1116.
[2] J.P. Perdew et al, Phys. Rev. Lett. 77 (1996) 3865–3868.
[3] M. Dion et al, Phys. Rev. Lett. 92 (2004) 246401.
[4] K. Lee et al, Phys. Rev. B 82 (2010) 081101.
[5] 4. Grenier R. et al, J. Phys. Chem. A, 119 (2015) 6897–6908.
[6] R.J. Maurer et al, J. Chem. Phys. 143 (2015) 102808.
[7] S. Gautier et al, Phys. Chem. Chem. Phys., 17 (2015) 28921.
Publication :
Masoud Shahrokhi, Céline Léonard. Quasi-particle energies and optical excitations of wurtzite BeO and its nanosheet. Journal of Alloys and Compounds, Elsevier, 2016, 682 (15), pp.254-262. <10.1016/ j.jallcom.2016.04.288>. <hal-01308725>