
Modeling of metal-hydrogen systems coupling DFT, CVM and Calphad methods
Advisors: Jean-Claude Crivello (ICMPE), Jean-Marc Joubert (ICMPE), Pierre Cénédèse (ICMPE)
Hydrogen absorption in the interstitial sites of metals is crucial for major issues such as alloy embrittlement or hydrogen storage for energy applications. This phenomenon modifies the physicochemical properties of the host metal and may lead to the formation of ordered MHy compounds called hydrides.
Within this framework, the Calphad modeling method (CALculation of PHAse Diagrams) is a relevant tool for understanding and predicting the behavior of metals and alloys in the presence of hydrogen. However, there is no Calphad database centered on hydrogen for calculating phase equilibria in multi-constituent systems (ternary, quaternary…).
The present thesis proposes to use a multi-scale modeling approach to study metal-hydrogen (M-H) binary systems, which are the first step in designing such a Calphad database.
First, systematic DFT (Density Functional Theory) calculations were carried out for 31 binary M-H systems considering 30 potential crystal structures, resulting in 30 × 31 = 930 hydrides, stable or metastable. This high throughput approach allowed in particular to determine the enthalpies of formation at 0 K, which represent important input data for the Calphad method. New hydrides that have never been experimentally observed could be predicted at high pressure (TaH2, ZrH3…).
Then, phonon calculations in the harmonic approximation were performed on the most stable hydrides. They allow, on the one hand, to correct the DFT calculated enthalpies of formation by considering the energy and entropy due to the atom vibrations, which are not negligible for the light hydrogen atom. On the other hand, a large-scale study focused on the modification of the free energy of formation due to hydrogen substitution by its isotopes, known as “isotopic effect”. Predictions were made on the nature of this effect as function of temperature.
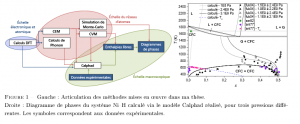
Moreover, the random insertion of hydrogen atoms in solid solution was studied using statistical thermodynamic methods: CVM (Cluster Variation Method) and Monte-Carlo simulation. These methods have been implemented in calculation codes, applied to face centered cubic (fcc) and body-centered cubic (bcc) metals. The input data are the interactions energies between nearest neighbor atoms. They are provided by the CEM (Cluster Expansion Method) coupled with DFT calculations. A comparative study of the Ni-H and Pd-H systems revealed the specificities of the thermodynamic behaviors of both solid interstitial solutions. Furthermore, dihydrogen pressure is an important parameter because many hydrides form only at very high pressure.
To improve the Calphad model accuracy at high pressure, the model of Lu et al. was applied to the condensed phases of the Ni-H, Rh-H and Mg-H systems. This model allows to determine the contribution to the free enthalpy due to the pressure force work. The input data may be both quasi-harmonic phonon calculation results and experimental data.
Finally, a comprehensive Calphad model of the Ni-H system was carried out by integrating the model of Lu et al. The DFT enthalpy of formation and the mixing enthalpy determined by CEM were used as input data, to complement the available experimental data.
Publications :
- Bourgeois, N., Crivello, J.-C., Cenedese, P., Paul-Boncour Valérie and Joubert, J. M., Vibration analysis of hydrogen, deuterium and tritium in metals: consequences on the isotope effect. Journal of Physics: Condensed Matter, (2018) doi: 10.1088/1361-648X/aad259.
- Bourgeois, N., Crivello, J.-C., Cenedese, P., Joubert, J.-M., A systematic first principle study of binary metal hydrides. ACS Comb. Sci. , 19 :8 (2017), 513-523.
- Bourgeois, N., Crivello, J.-C., Saengdeejing, A., Chen, Y., Cenedese, P., Joubert, J.-M., Thermo dynamic mo deling of the Ni-H system. J. Phys. Chem. C, 119 :43 (2015), 24546-24557.
- Crivello, J.-C., Souques, R., Breidi, A., Bourgeois, N., Joubert, J.-M., ZenGen, a tool to generate ordered configurations for systematic first-principles calculations : The Cr-Mo-Ni-Re system as a case study. Calphad, 51 (2015), 233-240