
A molecule can be adsorbed in two different ways on a surface. The first one is an adsorption relatively far from the surface which involves weak interactions (about ten meV), of van-der-Waals nature. This corresponds to the physisorption of the molecule. The second one, called chemisorption, involves breaking and creations of chemical bonds (a few eV) and takes place relatively close to the surface. We were mainly interested in this thesis in the modelling of the physisorption part of the potential for systems H2/metal where the metal is copper or silver. So far rather reliable methods exist to describe the chimisorption part of the potential while the modelling of the physisorption part remains a challenge nowadays for such systems. A better understanding of the adsorption of molecule is of major interest for heterogeneous catalysis because the chemical reactions often use this process to increase the rate of transformation or to improve the selectivity of the reaction, allowing large energy or material savings. Another possible application could be the storage of hydrogen on or into a surface.
First of all, during more than one year, we were interested in the physisorption of a hydrogen molecule on a (100) copper surface. Several approaches and ab-initio methods exist to calculate the interaction potential of H2/Cu system. Without taking into account the relaxation of the surface, such a system possesses six degrees of freedom described in Figure 1.
In a first step, we used the code VASP which works with plane waves basis set and considers the periodicity of the surface. The method of calculation used by this code is a relatively fast method called density functional theory (DFT). We could check several available functionals in a specific orientation of the hydrogen molecule above the surface and we selected the vdw-DF2 functional. This new functional has been designed to describe such types of interactions and provided in our case the best physisorption well depth according to experimental estimation.
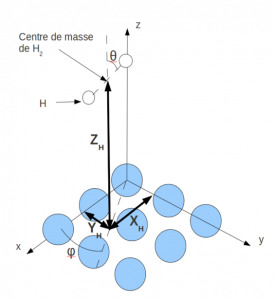
Figure 1
With this code and this functional, we obtained a 5D interaction potential for the hydrogen molecule over the surface: we verified that the H-H bond length does not vary in all the physisorption domain and remains at its equilibrium value. We observed that the rotation along the angle q is very weakly anisotropic, but the rotation along the helicopter angle j is almost isotropic. We also showed that there is almost no corrugation for the displacement of the hydrogen molecule over the surface. Consequently we could reduce the dimensionality to two (Z and q) coordinates for the subsequent study of the dynamics and the spectroscopy of the hydrogen molecule physisorbed on the copper surface.
Using the interaction potential obtained for the system H2/Cu(100), we calculated the ro-vibrational spectroscopy of the hydrogen molecule on the copper surface with a reduced Hamiltonian of the system (2D). However, the agreement between our calculated energy levels and those deduced from experiments were not good enough and we had to improve the quality of the interaction potential. For that, we used an Embedding approach of ONIOM type, which combines periodic calculations and more accurate local (cluster) model to correct from the missing delocalisation effects. The local model consists in a cluster of 22 atoms to represent the surface. The Embedding potential is expressed in the following way :
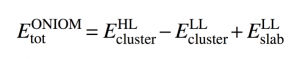 where the interaction energy of the H2 molecule with the cluster (22 atoms of copper forming 3 layers) was obtained firstly with the code VASP (and DFT calculations) and secondly with the code MOLPRO using the highly correlated CCSD(T) method. We then constructed the 2D Embedding potential (Z,q) and calculated the spectroscopy. With this corrected potential we could obtain much better results, namely very close to experience. This work on the system H2/Cu(100) led to the publication of an article [1] in February, 2015.
where the interaction energy of the H2 molecule with the cluster (22 atoms of copper forming 3 layers) was obtained firstly with the code VASP (and DFT calculations) and secondly with the code MOLPRO using the highly correlated CCSD(T) method. We then constructed the 2D Embedding potential (Z,q) and calculated the spectroscopy. With this corrected potential we could obtain much better results, namely very close to experience. This work on the system H2/Cu(100) led to the publication of an article [1] in February, 2015.
To have a more complete description of the ro-vibrational spectrum of the hydrogen molecule on the surface we also have to know the intensities of the transitions between the various energies levels. For that purpose, we calculated the 2D dipole moment function of the system (Z,q), within the Finite Field approach using the highly correlated CCSD(T) method. The transition probabilities between the energies levels have been determined with a first order time-dependent perturbation method, where the perturbation consists in a sinusoidal electric field. To obtain the quantities needed to simulate the spectra, we developed a quantum dynamics code based on the method of Time-Splitting operator. The evolution of a given initial state allowed us to obtain the matrix elements of the dipole moment which were found very close to experimental values (a second article is almost finished on this work).
We have also studied the physisorption of the hydrogen molecule on different types of surfaces: a (111) copper surface and (100) and (111) silver surfaces. The periodic calculations provided results which are comparable to those with the (100) copper surface. The continuation with the Embedding technique has been achieved with highly correlated methods, either multi-reference interaction of configurations (MRCI) or Coupled Cluster, with or without correction of basis set superposition error. For the copper (111), calculations are still in progress. For the surface of silver (100), the Embedding potential presents a very weak anisotropy for the rotation along the angle q, but gives rather good agreement with experiments for the transition energies between the various levels. For the surface of silver (111), the interaction potential calculated at the MRCI level seems to be better than the Embedding potential. The dipole moment function has been obtained directly from the MRCI calculation and is used to determine the dipole moment matrix elements within the quantum dynamics approach. It was found that for these last two systems the matrix elements are overestimated compared to experiment. To improve these results a new determination of the dipole moment function is in progress using the Finite Field method which is known to provide a better accuracy than direct MRCI calculation. We are writing an article on Ag/H2 systems.
Publication:
Bernard, C. Houriez, A. Mitrushchenkov, M. Guitou and G. Chambaud, (2015) Spectroscopy of H2 physisorbed on a Cu(100) surface, J. Chem. Phys., vol. 142, pp. 054703, (2015)